CytoNormPy - FCS files#
In this vignette, we showcase a typical analysis workflow using FCS files read from disk.
First, we import the necessary libraries
[1]:
import cytonormpy as cnp
import os
import pandas as pd
Metadata#
In order to tell cytonormpy, which data are references used for calculating the spline functions, we provide it with a metadata file as a pandas.DataFrame. Here, we read it from disk, but you can also create it on the fly using the pandas library.
[2]:
input_directory = "../_resources/"
output_directory = os.path.join(input_directory, "normalized")
if not os.path.exists(output_directory):
os.mkdir(output_directory)
metadata = pd.read_csv(os.path.join(input_directory, "metadata_sid.csv"))
metadata.head()
[2]:
| file_name | reference | batch | sample_ID | |
|---|---|---|---|---|
| 0 | Gates_PTLG021_Unstim_Control_1.fcs | ref | 1 | 1 |
| 1 | Gates_PTLG021_Unstim_Control_2.fcs | other | 1 | 2 |
| 2 | Gates_PTLG028_Unstim_Control_1.fcs | ref | 2 | 3 |
| 3 | Gates_PTLG028_Unstim_Control_2.fcs | other | 2 | 4 |
| 4 | Gates_PTLG034_Unstim_Control_1.fcs | ref | 3 | 5 |
Data setup#
We instantiate the cytonorm object and add a data transformer that will transform our data to the asinh space and the clusterer that will cluster the cells.
[3]:
cn = cnp.CytoNorm()
t = cnp.AsinhTransformer()
fs = cnp.FlowSOM(n_clusters=4)
cn.add_transformer(t)
cn.add_clusterer(fs)
The channels to be normalized are saved in a text file and will be passed to the run_fcs_data_setup() method.
Alternatively, valid arguments for the channels keyword are “markers” and “all”.
[4]:
coding_detectors = pd.read_csv(input_directory + "coding_detectors.txt", header=None)[0].tolist()
[5]:
cn.run_fcs_data_setup(
input_directory=input_directory,
metadata=metadata,
channels=coding_detectors,
output_directory=output_directory,
prefix="Norm",
)
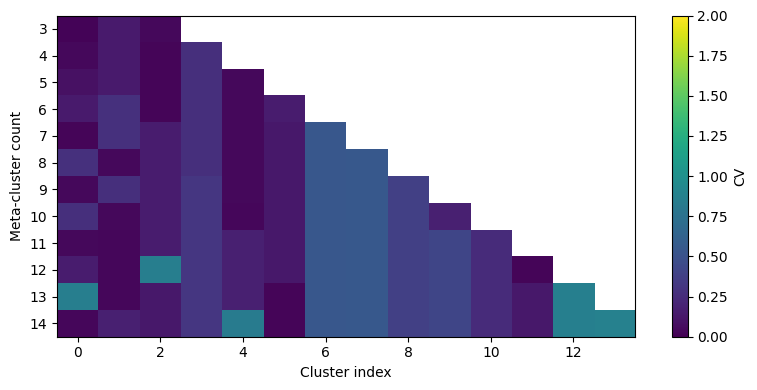
CV thresholding#
For clustering, it is important to visualize the distribution of files within one cluster. We have already added a FlowSOM Clusterer instance. the function ‘calculate_cluster_cvs’ will now calculate, for each metacluster number that we want to analyze, the cluster cv per sample.
We then visualize it via a waterfall plot as in the original CytoNorm implementation in R.
CytoNorm2.0: We can now use a different set of markers for clustering using the ‘markers’ parameter. If you want to use all markers, do not pass anything!
[6]:
markers_for_clustering = coding_detectors[4:15]
cn.calculate_cluster_cvs(n_metaclusters=list(range(3, 15)), markers=markers_for_clustering)
cnp.pl.cv_heatmap(cn, n_metaclusters=list(range(3, 15)), max_cv=2)
Clustering#
We run the FlowSOM clustering and pass a cluster_cv_threshold of 2. This value is used to evaluate if the distribution of files within one cluster is sufficient. A warning will be raised if that is not the case.
[7]:
cn.run_clustering(markers=markers_for_clustering, cluster_cv_threshold=2)
Calculation#
Finally, we calculate the quantiles per batch and cluster, calculate the spline functions and transform the expression values accordingly.
The data will automatically be saved to disk using the prefix Norm_. To change that prefix, pass the keyword prefix to the .run_fcs_data_setup() method above.
[8]:
cn.calculate_quantiles()
cn.calculate_splines(goal="batch_mean")
cn.normalize_data()
C:\Users\tarik\anaconda3\envs\cytonorm\lib\site-packages\cytonormpy\_cytonorm\_cytonorm.py:524: UserWarning: 23 cells detected in batch 1 for cluster 1. Skipping quantile calculation.
warnings.warn(warning_msg, UserWarning)
C:\Users\tarik\anaconda3\envs\cytonorm\lib\site-packages\cytonormpy\_cytonorm\_cytonorm.py:524: UserWarning: 32 cells detected in batch 1 for cluster 3. Skipping quantile calculation.
warnings.warn(warning_msg, UserWarning)
C:\Users\tarik\anaconda3\envs\cytonorm\lib\site-packages\cytonormpy\_cytonorm\_cytonorm.py:524: UserWarning: 6 cells detected in batch 1 for cluster 4. Skipping quantile calculation.
warnings.warn(warning_msg, UserWarning)
C:\Users\tarik\anaconda3\envs\cytonorm\lib\site-packages\cytonormpy\_cytonorm\_cytonorm.py:524: UserWarning: 41 cells detected in batch 1 for cluster 6. Skipping quantile calculation.
warnings.warn(warning_msg, UserWarning)
C:\Users\tarik\anaconda3\envs\cytonorm\lib\site-packages\cytonormpy\_cytonorm\_cytonorm.py:524: UserWarning: 15 cells detected in batch 1 for cluster 7. Skipping quantile calculation.
warnings.warn(warning_msg, UserWarning)
C:\Users\tarik\anaconda3\envs\cytonorm\lib\site-packages\cytonormpy\_cytonorm\_cytonorm.py:524: UserWarning: 5 cells detected in batch 1 for cluster 8. Skipping quantile calculation.
warnings.warn(warning_msg, UserWarning)
C:\Users\tarik\anaconda3\envs\cytonorm\lib\site-packages\cytonormpy\_cytonorm\_cytonorm.py:524: UserWarning: 3 cells detected in batch 1 for cluster 9. Skipping quantile calculation.
warnings.warn(warning_msg, UserWarning)
C:\Users\tarik\anaconda3\envs\cytonorm\lib\site-packages\cytonormpy\_cytonorm\_cytonorm.py:524: UserWarning: 17 cells detected in batch 1 for cluster 10. Skipping quantile calculation.
warnings.warn(warning_msg, UserWarning)
C:\Users\tarik\anaconda3\envs\cytonorm\lib\site-packages\cytonormpy\_cytonorm\_cytonorm.py:524: UserWarning: 2 cells detected in batch 1 for cluster 12. Skipping quantile calculation.
warnings.warn(warning_msg, UserWarning)
C:\Users\tarik\anaconda3\envs\cytonorm\lib\site-packages\cytonormpy\_cytonorm\_cytonorm.py:524: UserWarning: 9 cells detected in batch 1 for cluster 13. Skipping quantile calculation.
warnings.warn(warning_msg, UserWarning)
C:\Users\tarik\anaconda3\envs\cytonorm\lib\site-packages\cytonormpy\_cytonorm\_cytonorm.py:524: UserWarning: 14 cells detected in batch 2 for cluster 1. Skipping quantile calculation.
warnings.warn(warning_msg, UserWarning)
C:\Users\tarik\anaconda3\envs\cytonorm\lib\site-packages\cytonormpy\_cytonorm\_cytonorm.py:524: UserWarning: 43 cells detected in batch 2 for cluster 3. Skipping quantile calculation.
warnings.warn(warning_msg, UserWarning)
C:\Users\tarik\anaconda3\envs\cytonorm\lib\site-packages\cytonormpy\_cytonorm\_cytonorm.py:524: UserWarning: 8 cells detected in batch 2 for cluster 4. Skipping quantile calculation.
warnings.warn(warning_msg, UserWarning)
C:\Users\tarik\anaconda3\envs\cytonorm\lib\site-packages\cytonormpy\_cytonorm\_cytonorm.py:524: UserWarning: 7 cells detected in batch 2 for cluster 7. Skipping quantile calculation.
warnings.warn(warning_msg, UserWarning)
C:\Users\tarik\anaconda3\envs\cytonorm\lib\site-packages\cytonormpy\_cytonorm\_cytonorm.py:524: UserWarning: 10 cells detected in batch 2 for cluster 8. Skipping quantile calculation.
warnings.warn(warning_msg, UserWarning)
C:\Users\tarik\anaconda3\envs\cytonorm\lib\site-packages\cytonormpy\_cytonorm\_cytonorm.py:524: UserWarning: 1 cells detected in batch 2 for cluster 9. Skipping quantile calculation.
warnings.warn(warning_msg, UserWarning)
C:\Users\tarik\anaconda3\envs\cytonorm\lib\site-packages\cytonormpy\_cytonorm\_cytonorm.py:524: UserWarning: 14 cells detected in batch 2 for cluster 10. Skipping quantile calculation.
warnings.warn(warning_msg, UserWarning)
C:\Users\tarik\anaconda3\envs\cytonorm\lib\site-packages\cytonormpy\_cytonorm\_cytonorm.py:524: UserWarning: 49 cells detected in batch 2 for cluster 11. Skipping quantile calculation.
warnings.warn(warning_msg, UserWarning)
C:\Users\tarik\anaconda3\envs\cytonorm\lib\site-packages\cytonormpy\_cytonorm\_cytonorm.py:524: UserWarning: 1 cells detected in batch 2 for cluster 12. Skipping quantile calculation.
warnings.warn(warning_msg, UserWarning)
C:\Users\tarik\anaconda3\envs\cytonorm\lib\site-packages\cytonormpy\_cytonorm\_cytonorm.py:524: UserWarning: 3 cells detected in batch 2 for cluster 13. Skipping quantile calculation.
warnings.warn(warning_msg, UserWarning)
C:\Users\tarik\anaconda3\envs\cytonorm\lib\site-packages\cytonormpy\_cytonorm\_cytonorm.py:524: UserWarning: 11 cells detected in batch 3 for cluster 1. Skipping quantile calculation.
warnings.warn(warning_msg, UserWarning)
C:\Users\tarik\anaconda3\envs\cytonorm\lib\site-packages\cytonormpy\_cytonorm\_cytonorm.py:524: UserWarning: 12 cells detected in batch 3 for cluster 4. Skipping quantile calculation.
warnings.warn(warning_msg, UserWarning)
C:\Users\tarik\anaconda3\envs\cytonorm\lib\site-packages\cytonormpy\_cytonorm\_cytonorm.py:524: UserWarning: 47 cells detected in batch 3 for cluster 6. Skipping quantile calculation.
warnings.warn(warning_msg, UserWarning)
C:\Users\tarik\anaconda3\envs\cytonorm\lib\site-packages\cytonormpy\_cytonorm\_cytonorm.py:524: UserWarning: 24 cells detected in batch 3 for cluster 7. Skipping quantile calculation.
warnings.warn(warning_msg, UserWarning)
C:\Users\tarik\anaconda3\envs\cytonorm\lib\site-packages\cytonormpy\_cytonorm\_cytonorm.py:524: UserWarning: 6 cells detected in batch 3 for cluster 8. Skipping quantile calculation.
warnings.warn(warning_msg, UserWarning)
C:\Users\tarik\anaconda3\envs\cytonorm\lib\site-packages\cytonormpy\_cytonorm\_cytonorm.py:524: UserWarning: 7 cells detected in batch 3 for cluster 9. Skipping quantile calculation.
warnings.warn(warning_msg, UserWarning)
C:\Users\tarik\anaconda3\envs\cytonorm\lib\site-packages\cytonormpy\_cytonorm\_cytonorm.py:524: UserWarning: 23 cells detected in batch 3 for cluster 10. Skipping quantile calculation.
warnings.warn(warning_msg, UserWarning)
C:\Users\tarik\anaconda3\envs\cytonorm\lib\site-packages\cytonormpy\_cytonorm\_cytonorm.py:524: UserWarning: 40 cells detected in batch 3 for cluster 11. Skipping quantile calculation.
warnings.warn(warning_msg, UserWarning)
C:\Users\tarik\anaconda3\envs\cytonorm\lib\site-packages\cytonormpy\_cytonorm\_cytonorm.py:524: UserWarning: 7 cells detected in batch 3 for cluster 12. Skipping quantile calculation.
warnings.warn(warning_msg, UserWarning)
C:\Users\tarik\anaconda3\envs\cytonorm\lib\site-packages\cytonormpy\_cytonorm\_cytonorm.py:524: UserWarning: 11 cells detected in batch 3 for cluster 13. Skipping quantile calculation.
warnings.warn(warning_msg, UserWarning)
C:\Users\tarik\anaconda3\envs\cytonorm\lib\site-packages\cytonormpy\_normalization\_quantile_calc.py:274: RuntimeWarning: Mean of empty slice
self.distrib = mean_func(expr_quantiles._expr_quantiles, axis=self._batch_axis)
normalized file Gates_PTLG028_Unstim_Control_1.fcs
normalized file Gates_PTLG021_Unstim_Control_1.fcs
normalized file Gates_PTLG034_Unstim_Control_1.fcs
normalized file Gates_PTLG028_Unstim_Control_2.fcs
normalized file Gates_PTLG021_Unstim_Control_2.fcs
normalized file Gates_PTLG034_Unstim_Control_2.fcs
C:\Users\tarik\anaconda3\envs\cytonorm\lib\site-packages\cytonormpy\_dataset\_dataset.py:376: RuntimeWarning: overflow encountered in cast
orig_events[:, channel_indices] = inv_transformed.values
In order to use the cytonorm object on new data, simply pass the filenames and the batch information. You can either pass a single filename or provide a list.
[9]:
cn.normalize_data(file_names="Gates_PTLG034_Unstim_Control_2_dup.fcs", batches=3)
normalized file Gates_PTLG034_Unstim_Control_2_dup.fcs